
Oamenii de ştiinţă fac noi progrese în cercetarea unei tulburări genetice rare, sindromul Ehlers-Danlos (SED/EDS) - boala corpului îndoit - care determină organismul să producă ţesut conjunctiv defect şi duce la simptome debilitante de hipermobilitate, instabilitate articulară şi fragilitate a pielii.
Sindromul Ehlers-Danlos este o boală genetică rară şi cuprinde un grup heterogen, din punct de vedere clinic şi genetic, de afecţiuni ale ţesutului conjunctiv caracterizate prin hipermobilitate, tegument hiperelastic şi fragilitate tisulară. Deşi boala afectează ambele sexe, apare mai des la femei şi nu este vindecabilă.
Această tulburare genetică rară determină organismul să producă ţesut conjunctiv defect, iar ţesutul conjunctiv este peste tot - în tendoane, ligamente, piele, inimă, sistemul digestiv, ochi şi gingii.
Mulţi pacienţi cu SED petrec ani de zile până să fie diagnosticaţi corect; alţii au simţit că au fost neglijaţi şi izolaţi de medici.
Linda Bluestein, un medic din Colorado, specializat în SED şi alte afecţiuni de hipermobilitate suferă şi ea de SEDh (hEDS).
Primele dureri articulare şi migrene i-au apărut la vârsta de 16 ani când a şi suferit prima operaţie ortopedică, dar a primit diagnosticul de SEDh abia la vârsta de 47 de ani, adică mai mult de 30 de ani mai târziu.
„I-am spus medicului meu de multe ori, „e ceva în neregulă cu mine, nu mă vindec bine, mă rănesc mai uşor decât alţii”, spune ea. „Şi pur şi simplu nu a ascultat niciodată, niciodată”.
De ce, pentru atât de mulţi pacienţi, durează atât de mult până să fie diagnosticaţi?
Mult mai multe femei decât bărbaţi sunt diagnosticaţi cu SED, ceea ce ar putea explica neglijarea diagnosticării bolii, deoarece profesia medicală are o istorie lungă în care plângerile de sănătate făcute de femei sunt ignorate, spune dr. Bluestein.
Un studiu din 2009, realizat de Organizaţia Europeană pentru Boli Rare, a chestionat 414 familii de pacienţi cu SED din cinci ţări şi a constatat că timpul mediu până la diagnosticarea SED a fost de patru ani pentru bărbaţi, dar de 16 ani pentru femei.
Raportul afirmă că femeile cu SED tind să fie „diagnosticate mai târziu, deoarece durerea şi hipotonia acestora (tonus muscular slab) nu sunt considerate simptome fizice, ci mai degrabă simptome psihologice sau plângeri obişnuite”.
„Avem tendinţa de a nu fi luate în seamă mult mai uşor”, spune dr. Bluestein. „Oamenii ajung la concluzia că suntem femei histrionice”, spune ea.
Anxietatea este foarte frecventă în rândul pacienţilor cu probleme de hipermobilitate, explică dr. Bluestein, ceea ce poate complica lucrurile. „Când persoanele cu anxietate se prezintă la un medic, anxietatea iese în evidenţă şi poate fi copleşitoare, astfel încât medicul aproape că nu poate vedea nimic altceva”.
Acest lucru poate spori şi mai mult anxietatea pacientului „pentru că oamenii nu ne validează simptomele şi apoi începem să ne îndoim de noi înşine”, spune medicul.
Ea a explicat faptul că pacienţii nediagnosticaţi merg deseori să consulte un neurolog pentru migrenele lor, un reumatolog pentru durerile articulare, un cardiolog pentru palpitaţii, un gastroenterolog pentru problemele digestive şi un urolog pentru simptomele vezicii urinare.
Fiecare medic se concentrează pe simptomele care se încadrează în specialitatea sa, dar nu ia în considerare celelalte afecţiuni. „Nicăieri pe parcurs nu realizează cineva că există anumite afecţiuni care ar putea lega toate aceste simptome împreună şi ar putea explica totul”, spune dr. Bluestein.
Studiul privind bolile rare din 2009 a constatat că, în timpul căutării unui diagnostic, 58% dintre pacienţii cu SED au consultat mai mult de cinci medici, iar 20% au consultat mai mult de 20.
Consecinţele de a nu fi diagnosticat ani de zile pot fi devastatoare, subliniază medicul.
Pacienţii cu SEDh îşi spun în mod frecvent „zebre medicale” pentru a evoca raritatea bolii şi dungile de vergeturi care apar pe piele şi care reprezintă o caracteristică comună a SED.
Potrivit unor statistici, sindromul afectează 1:2.500–1:5.000 de indivizi, dar specialiştii spun că incidenţa bolii ar putea fi mult mai mare şi, deşi tulburările de hipermobilitate pot fi debilitante, timpul mediu până la diagnosticare de la debutul simptomelor este de 10 până la 12 ani, potrivit Societăţii Ehlers-Danlos.
Cercetările limitate care au fost efectuate asupra prevalenţei SEDh sugerează că numărul real de cazuri este „mult, mult mai mare” decât atât, spune dr. Linda Bluestein care a tratat pacienţi cu SEDh care au căutat un diagnostic timp de zeci de ani.
Medicul a indicat un studiu din 2019 realizat în Ţara Galilor – o ţară de 3,1 milioane de oameni. O examinare a asistenţei medicale primare şi a dosarelor spitaliceşti din 1990 până în 2017 a constatat că una din 500 de persoane din această ţară are fie SEDh, fie sindrom de hipermobilitate articulară, o afecţiune similară cu un set uşor diferit de simptome. Se crede însă că există încă o subestimare a numărului persoanelor care ar putea suferi de această afecţiune.
Potrivit societăţii Ehlers-Danlos, ar trebui făcute mai multe studii asupra populaţiei pentru a oferi o imagine mai precisă a incidenţei acestei afecţiuni în lume.
Sindromul pare să fie cauzat de un defect în structura, producerea sau prelucrarea colagenului sau a proteinelor care interacţionează cu colagenul, cum ar fi mutaţii în genele COL5A sau COL3A. Ca urmare, apar modificări în ţesutul conjunctiv; fragilitatea pielii şi instabilitatea articulară sunt rezultatul cantităţii reduse ori calităţii defectuoase a colagenului.
Caracteristicile clinice ale SED au fost descrise pentru prima dată de Hipocrate (400 î.Hr.). Sindromul este numit după medicii Edvard Ehlers (Danemarca) şi Henri-Alexandre Danlos (Franţa), care au descris afecţiunea la începutul secolului 20.
Există 13 tipuri de sindrom Ehlers-Danlos conform organizaţiei de cercetare şi societăţii Ehlers-Danlos. Cele mai multe tipuri sunt foarte rare şi pot fi diagnosticate folosind teste genetice. Cu toate acestea, genele care cauzează SED hipermobil (SEDh) – cea mai comună formă, reprezentând aproximativ 90% din cazuri – sunt necunoscute, aşa că diagnosticul se bazează pe o listă de verificare a simptomelor.
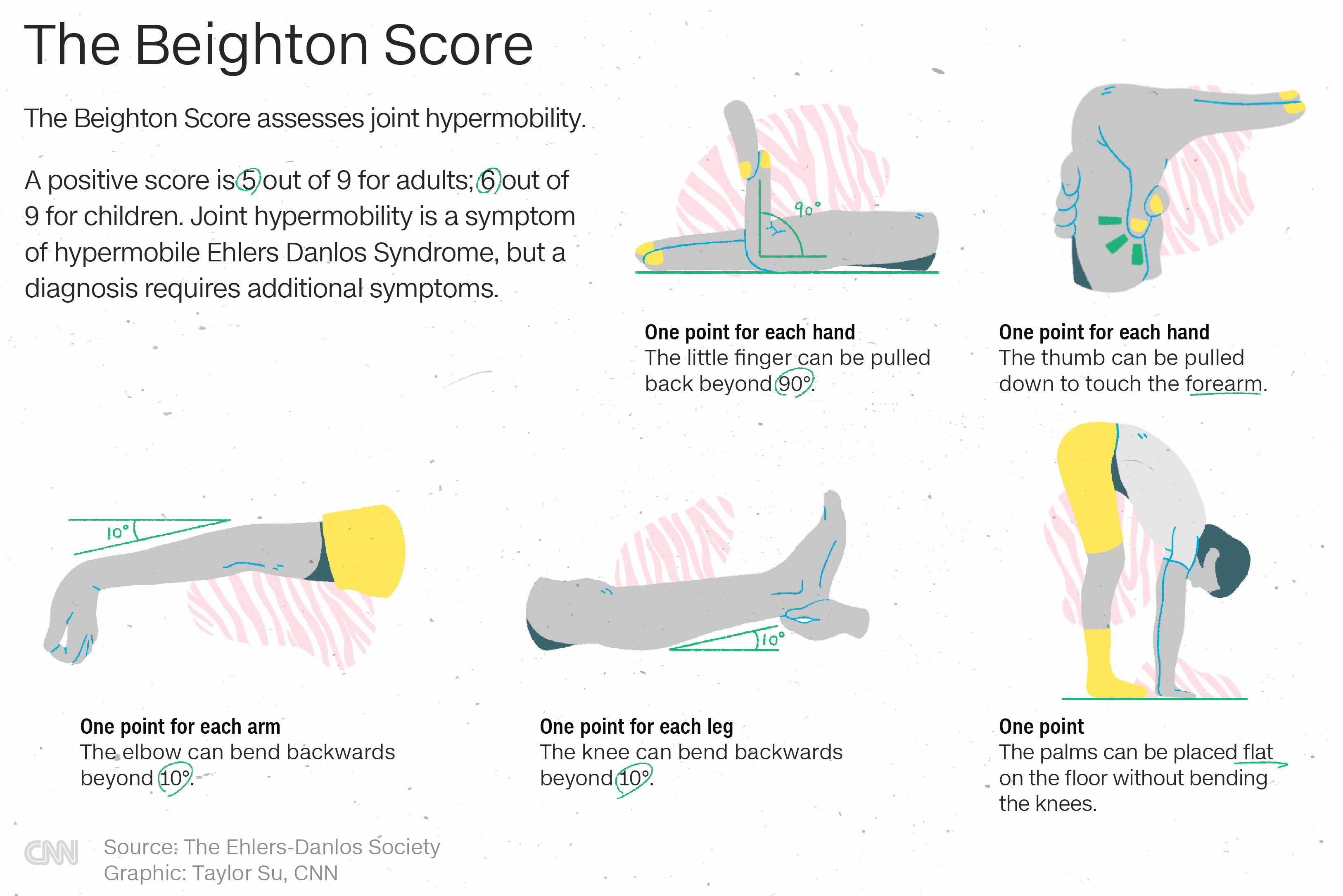
Pentru măsurarea hipermobilităţii articulare există un test de evaluare cunoscut sub numele de Scorul Beighton.
Melissa Dickinson, psihoterapeut din Atlanta, Georgia, spune că a experimentat simptomele unei „boli misterioase” încă din copilărie. Apoi, în 2013, când se afla în luna de miere în Mexic, relativ sănătoasă, s-a întors din vacanţă cu handicap şi cu gâtul luxat.
În timp ce se afla în vacanţă, Dickinson spune că a suferit o indigestie alimentară şi i s-a prescris ciprofloxacină, un antibiotic care poate prezenta un risc grav pentru persoanele cu SED. Medicamentul i-a declanşat leziuni semnificative ale nervilor, probleme digestive care aproape au făcut-o să orbească, deoarece corpul ei nu absorbea nutrienţi, şi a pus-o într-un scaun cu rotile, spune ea.
Dickinson, care a fost diagnosticată într-un final cu SEDh în 2014, spune că luând medicamente greşite i-a produs „distrugere din cap până în picioare”. Acum că primeşte tratament, poate merge cu mijloace de sprijin, şi, în cea mai mare parte a timpului corpul său trebuie să aibă un sprijin constant pentru a funcţiona.
Lara Bloom, preşedinte şi CEO al Societăţii Ehlers-Danlos, care ea însăşi are SEDh, spune că mulţi pacienţi au „PTSD din cauze medicale”.
Uneori, eşecul de a diagnostica SED a dus la îndepărtarea copiilor din grija propriilor părinţi.
În 2010, americanii Rana Tyson şi soţul ei Chad au fost acuzaţi în mod fals că le-au făcut rău fiicelor lor gemene în vârstă de 4 săptămâni, care aveau fracturi inexplicabile la picioare. Alături de sora lor mai mare, fetiţele au fost luate de autorităţile de stat din Texas şi trimise să locuiască la rude.
Cinci luni mai târziu, un genetician a identificat faptul că gemenele aveau o tulburare a ţesutului conjunctiv şi, ulterior, au fost diagnosticate cu SED şi o deficienţă de vitamina D.
Bloom spune că şi alţi părinţi cu copii cu SED au fost acuzaţi în mod greşit de „boală artificială sau indusă (FII)” – o formă rară de abuz, cunoscută anterior ca sindromul Munchausen by proxy, în care un părinte sau îngrijitor provoacă în mod deliberat simptome sau încearcă să convingă medicii că un copil sănătos este bolnav.
Unii pacienţi diagnosticaţi cu probleme digestive severe legate de SEDh, au fost iniţial asociaţi cu tulburări de alimentaţie (eating disorder).
În cazul SEDh, un prim pas crucial este acela de a afla ce cauzează această boală
Cortney Gensemer, om de ştiinţă la departamentul de medicină regenerativă şi biologie celulară a universităţii medicale din Carolina de Sud, încearcă să rezolve acest mister. Ea a studiat o mutaţie genetică despre care crede că provoacă SEDh - rezultatele studiului sunt în prezent în curs de evaluare inter pares.
Şi Gensemer a fost diagnosticată cu SEDh în adolescenţă. Ea spune că boala îi afectează fiecare aspect al muncii sale. Privitul la microscop poate fi uneori deosebit de dureros, gâtul ei fiind instabil din cauza SEDh. Cercetătoarea a necesitat şuruburi metalice în unele dintre vertebrele gâtului pentru a le fuziona.
Laboratorul său a fost dotat cu echipamente speciale, inclusiv uşi cu senzori de mişcare (uşile standard ale laboratorului fiind foarte grele), scaune reglabile şi pipete ergonomice care sunt uşor de mânuit „Dacă nu aş avea toate aceste lucruri, nu cred că aş fi în stare să-mi duc la capăt cercetările”, spune Gensemer.
Pentru a găsi genele care provoacă SEDh, cercetătoarea a prelevat probe de ADN dintr-o familie numeroasă care suferă de această afecţiune, cu cazuri care se întind pe patru generaţii, şi a căutat o mutaţie care apare doar la rudele care suferă de această boală. Ea a identificat o „genă candidat puternică” pe care a introdus-o la şoareci folosind instrumente de editare a genelor.
Gensemer şi mentorul său în cercetare, dr. Russell Norris, şeful laboratorului de cercetare, au descoperit că şoarecii SEDh aveau ţesuturi semnificativ mai laxe şi cozi mai moi decât rozătoarele obişnuite. „Poţi face cu uşurinţă un nod în coada şoarecelui mutant. Cu o coadă normală de şoarece, o poţi (doar) îndoi într-un cerc”, spune Gensemer.
Gena pe care Gensemer şi Norris au descoperit-o nu va putea explica toate cazurile de SEDh, dar cercetătorii cred că în cele din urmă vor fi identificate mai multe gene, iar SEDh va putea fi împărţită în diferite subtipuri, care ar putea explica de ce diferiţi pacienţi au simptome diferite.
În mod esenţial, dacă informaţiile genetice vor face lumină asupra modului în care ţesutul conjunctiv este „dat peste cap”, acest lucru ar putea duce la dezvoltarea unor potenţiale tratamente eficiente, spune Gensemer.
Fundaţia de cercetare a societăţii Ehlers-Danlos caută, de asemenea, să identifice genele, şi eventual şi markeri de sânge, lucrând cu o echipă de experţi pentru a secvenţia şi analiza ADN-ul a 1.000 de pacienţi cu SEDh din întreaga lume.
Candidatul la doctorat Sabeeha Malek, de la universitatea Warwick, din Marea Britanie, o altă cercetătoare care suferă de SEDh, a propus drept cauză pentru această afecţiune o defecţiune a modului în care colagenul se leagă de membranele celulare din ţesutul conjunctiv. Dacă va putea dovedi acest lucru cercetările ei ar putea duce la dezvoltarea unui test de biopsie a pielii care ar putea identifica formele acestei boli rare şi atât de puţin cunoscute.




