
O terapie genică experimentală a restabilit auzul şi a arătat semne de îmbunătăţire a vederii la animale şi organoizi.
Cercetătorii de la facultatea de medicină a universităţii Harvard (HMS) au făcut încă un pas decisiv în eforturile de a dezvolta o terapie genică pentru persoanele cu sindrom Usher de tip 1F, o afecţiune rară care provoacă surditate şi duce progresiv orbire.
Sindromul Usher este o boală genetică (moştenită) rară, caracterizată prin pierderea parţială sau totală a auzului şi a vederii, care se agravează în timp. Boala afectează în jur de 4 pana la 17 din 100.000 de persoane la nivel mondial.
Persoanele cu sindrom Usher de tip 1F se nasc fără auz sau simţul echilibrului şi îşi pierd treptat vederea.
O nouă modalitate de livrare a unei versiuni corectate a genei defecte care provoacă sindromul Usher - PCDH15 - a restabilit auzul la modelele de şoareci şi a arătat potenţial în organoizii retinieni şi la primatele neumane pentru îmbunătăţirea vederii, raportează echipa în Journal of Clinical Investigation.
Este a doua terapie genică experimentală pentru sindromul Usher dezvoltată de laboratorul Harvard de ştiinţe medicale translaţionale, din cadrul Institutului Blavatnik.
Cercetările anterioare au arătat că o strategie diferită de administrare a genelor a restabilit auzul la şoareci.
Noua metodă oferă o a doua opţiune în cazul în care prima abordare se dovedeşte nesigură sau ineficientă atunci când va fi testată la om.
„Fără un studiu clinic la om, nu putem şti dacă prima noastră terapie genică restabileşte funcţia normală”, a declarat David Corey, profesor de ştiinţe medicale translaţionale în cadrul Institutului Blavatnik de la HMS, autorul principal al studiului, într-un comunicat al universităţii.
„Această nouă strategie ne oferă o rezervă în cazul în care prima terapie nu funcţionează. S-ar putea chiar să se dovedească a fi mai bună decât prima, odată testată pe pacienţi”, a precizat cercetătorul de la Harvard.
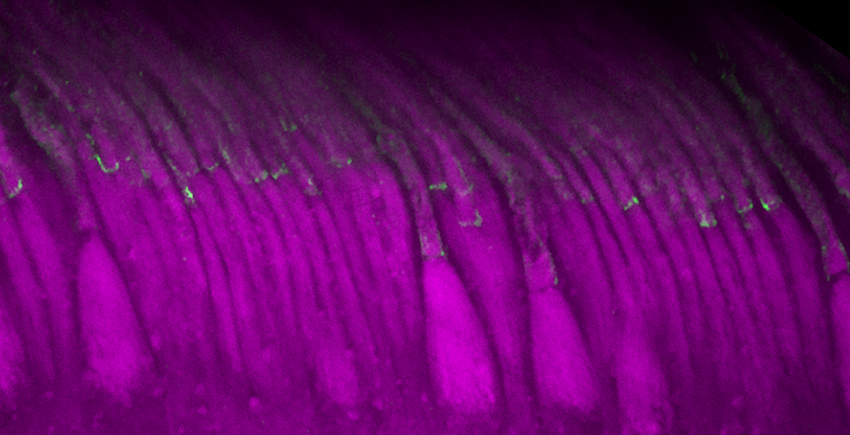
O terapie genică experimentală a crescut nivelul unei proteine cheie, protocadherina-15, prezentată în verde, în celulele sensibile la lumină din retina primatelor neumane. Imagine: Maryna Ivanchenko, Universitatea Harvard, facultatea de medicină, 15 noiembrie 2024
Mai multe strategii, mai multe şanse de vindecare
Provocarea principală în conceperea unei terapii cu gena PCDH15 este că aceasta este prea mare pentru a încăpea într-un virus adeno-asociat (AAV) - cel mai comun şi mai sigur vehicul utilizat pentru a transporta gene în celulele ţintă.
Strategia anterioară a echipei a implicat reducerea PCDH15 la o genă mini care să poată încăpea într-un AAV.
Noua strategie presupune segmentarea genei complete în două, inserarea fiecărei jumătăţi într-un AAV şi trimiterea AAV-urilor la urechea internă sau la ochi. Acolo, jumătăţile se reunesc şi încep să instruiască celulele să producă corect proteina codificată de genă, protocadherina-15. Abordarea a restabilit auzul şi echilibrul la şoareci.
Modelele existente de şoareci nu prezintă tipul de pierdere a vederii observat în sindromul Usher de tip 1F, astfel încât echipa nu a putut studia dacă vederea acestora s-a îmbunătăţit. Cu toate acestea, terapia Dual-AAV a crescut nivelurile de protocadherină-15 în celulele sensibile la lumină ale organoizilor retinieni umani şi ale retinei primatelor neumane. De asemenea, proteina a migrat la locul potrivit în aceste celule. Ambele rezultate sugerează că strategia de tratament ar putea acţiona într-o zi pentru a păstra vederea pacienţilor.
„Aceste rezultate sunt deosebit de importante deoarece, în timp ce implanturile cohleare pot aborda pierderea auzului la pacienţii umani, în prezent nu există tratamente pentru disfuncţia vederii asociată cu sindromul Usher”, a declarat primul autor Maryna Ivanchenko, instructor HMS în neurobiologie şi oftalmolog.
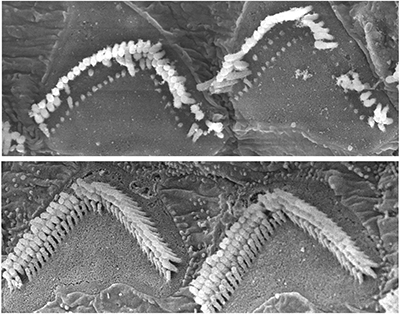
Modelele de şoareci cu sindrom Usher de tip 1F prezintă fascicule anormale de celule ciliate în urechea internă, sus. Terapia Dual-AAV a îmbunătăţit structura şi funcţia celulelor la şoareci, jos. Imagine: Maryna Ivanchenko, Universitatea Harvard, facultatea de medicină, 15 noiembrie 2024




